Background: Hyperphosphatasia with Impaired Intellectual Development Syndrome (HPMRS, Mabry syndrome) is a rare autosomal recessive disorder caused by mutations in genes responsible for glycosylphosphatidylinositol (GPI)-anchor biosynthesis. It is characterized by persistently elevated serum alkaline phosphatase (ALP), intellectual disability, seizures, dysmorphic features, and skeletal abnormalities. Case Presentation: We report the case of an 18-month-old Iraqi girl with genetically confirmed HPMRS-1 (PIGV mutation). She presented with refractory generalized tonic–clonic seizures, cortical blindness, hypotonia, global developmental delay, and distinct craniofacial and skeletal dysmorphism. Laboratory investigations showed persistently elevated ALP levels (2–20× above normal). Brain MRI demonstrated cerebral atrophy. Whole-exome sequencing confirmed a pathogenic PIGV mutation consistent with HPMRS-1. Despite treatment with multiple antiepileptic drugs including valproate, brivaracetam, lacosamide, and pyridoxine, seizures remained poorly controlled. Conclusion: This is the first genetically confirmed case of HPMRS reported from Iraq. The case highlights the importance of considering HPMRS in infants presenting with developmental delay, refractory seizures, and unexplained hyperphosphatasemia. Early diagnosis is essential for genetic counseling, supportive management, and avoidance of unnecessary investigations.
Keywords
HPMRS
PIGV
ALP
Hyperphosphatasia
Autosomal
INTRODUCTION
Hyperphosphatasia with Impaired Intellectual Development Syndrome (HPMRS), first described by Mabry et al. [1], represents a group of rare inherited disorders caused by defects in GPI-anchor biosynthesis [1]. Mutations in PIGV, PIGO, PGAP2, PGAP3, PIGW, PIGY, and PIGL disrupt the attachment of ALP and other GPI-anchored proteins to the cell surface, leading to persistent hyperphosphatasemia [2-4].
Clinically, HPMRS is characterized by moderate-to-severe intellectual disability, seizures, hypotonia, distinctive facial features, brachytelephalangy, feeding difficulties, and sometimes congenital heart or gastrointestinal anomalies [5-7]. The severity varies according to the affected gene. We report the first genetically confirmed case of HPMRS from Iraq, emphasizing its clinical, biochemical, and genetic findings to raise awareness of this underrecognized condition.
Case Presentation
Patient Information
Name: Lababa Hassan Ali
Age: 18 months
Address: Taza, Kirkuk, Iraq
Date of Admission: 29 July 2025
Date of History and Examination: 31 July 2025
Chief Complaint
Recurrent abnormal body movements manifested as generalized tonic–clonic seizures associated with eye rolling, frothy secretions, and loss of consciousness lasting approximately four minutes, occurring three times daily.
History of Present Illness
The patient was a preterm infant diagnosed with genetically confirmed HPMRS-1 (PIGV mutation) at 12
months. She had been on regular neurological follow-up and received valproate, brivaracetam, lacosamide, and pyridoxine, yet seizures persisted. Two weeks before admission, she was hospitalized for chest infection and seizure exacerbation; vitamin B6 was added, but seizure frequency increased without fever, vomiting, or trauma.
Past Medical History
Feeding difficulties and irritability since birth.
At 5 months: recurrent infantile spasms evolving into generalized seizures.
At 1 year: Whole-exome sequencing confirmed PIGV mutation.
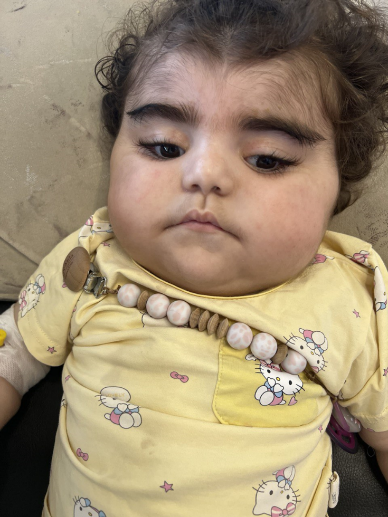
Figure 1: Facial features of the patient with HPMRS (Mabry syndrome) Frontal view showing characteristic craniofacial dysmorphism including a broad forehead, thick eyebrows, long eyelashes, and a flat nasal bridge
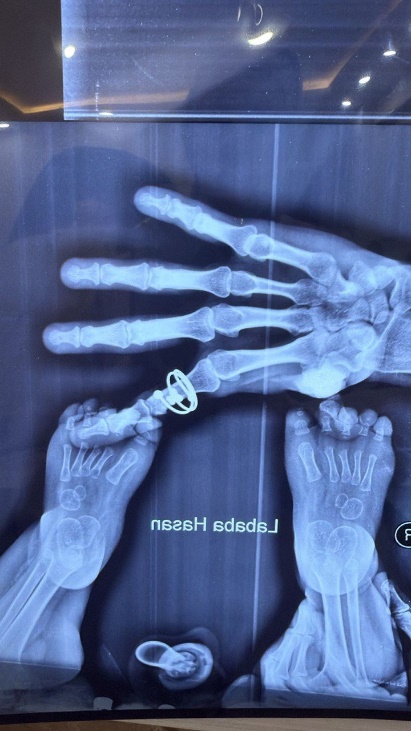
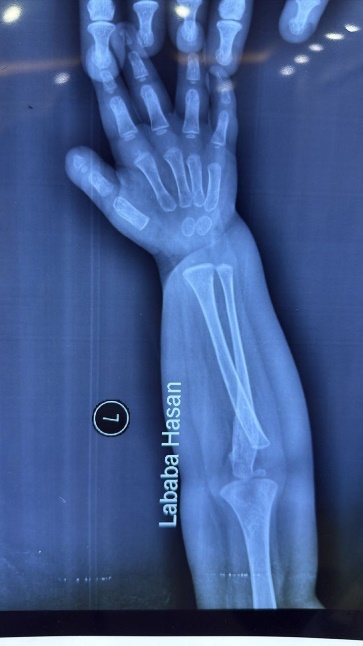
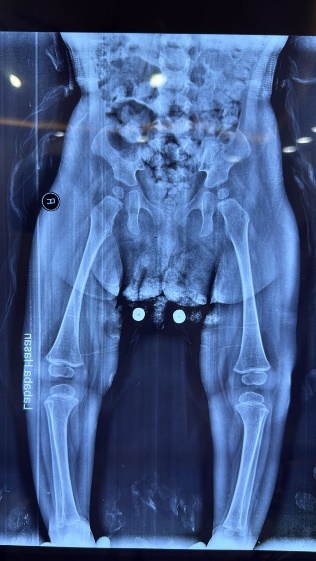
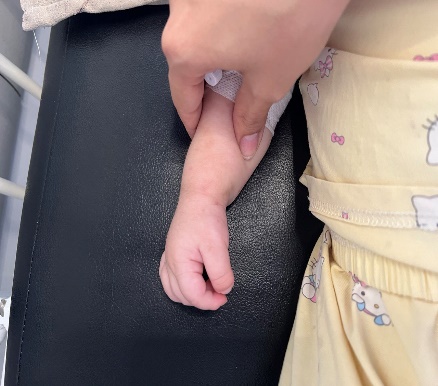
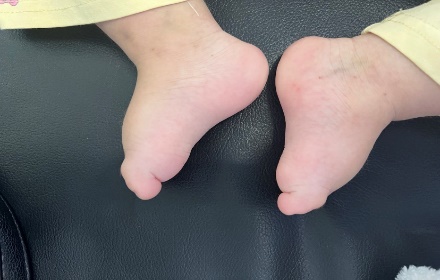
Figure 2: Hands and feet deformities
Short, broad hands and feet with clenched fingers and bilateral clubfoot, consistent with skeletal anomalies described in HPMR
Multiple hospitalizations for seizure control.
Congenital heart disease (ASD) repaired surgically at 1 year.
Developmental History
Severe global developmental delay: bedridden, unable to sit, crawl, or stand; no purposeful grasp, speech, or social interaction.
Family History
First child of non-consanguineous, healthy parents.
No family history of epilepsy or genetic disorders.
Anthropometry: Weight 10 kg (25th centile), length 77 cm (10th), OFC 47 cm (75th).
Other systemic examinations were unremarkable.
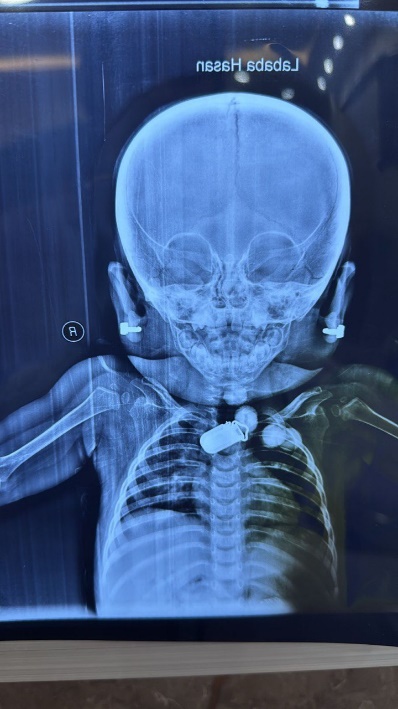
Figure 3: Brain MRI of the patient
Axial T2-weighted MRI demonstrating diffuse cerebral atrophy with widened sulci and ventricular dilatation
Figure 4: Pedigree and genetic analysis
Family pedigree and Sanger sequencing confirming the pathogenic homozygous PIGV mutation in the proband; both parents are heterozygous carriers
Figure 5: Comprehensive management plan
Summary of the multidisciplinary management including antiepileptic regimen (valproate, brivaracetam, lacosamide, pyridoxine), physiotherapy, nutritional support, and genetic counseling
Figure 6: Genetic testing report – Farabi Medical Laboratory Page displaying methodology and limitations of the supplemental gene sequencing confirming GPI-anchor pathway mutation analysis performed in Erbil, Iraq
Examination Findings
General: Unresponsive, afebrile; no pallor or cyanosis.
Craniofacial: Broad forehead, thick eyebrows, long eyelashes, flat nasal bridge.
Skeletal: Short broad hands/feet, clenched fists, bilateral clubfoot.
Neurological: Cortical blindness, generalized hypotonia, muscle power 3/6, decreased reflexes.
Anthropometry: Weight 10 kg (25th centile), length 77 cm (10th), OFC 47 cm (75th).
Other systemic examinations were unremarkable.
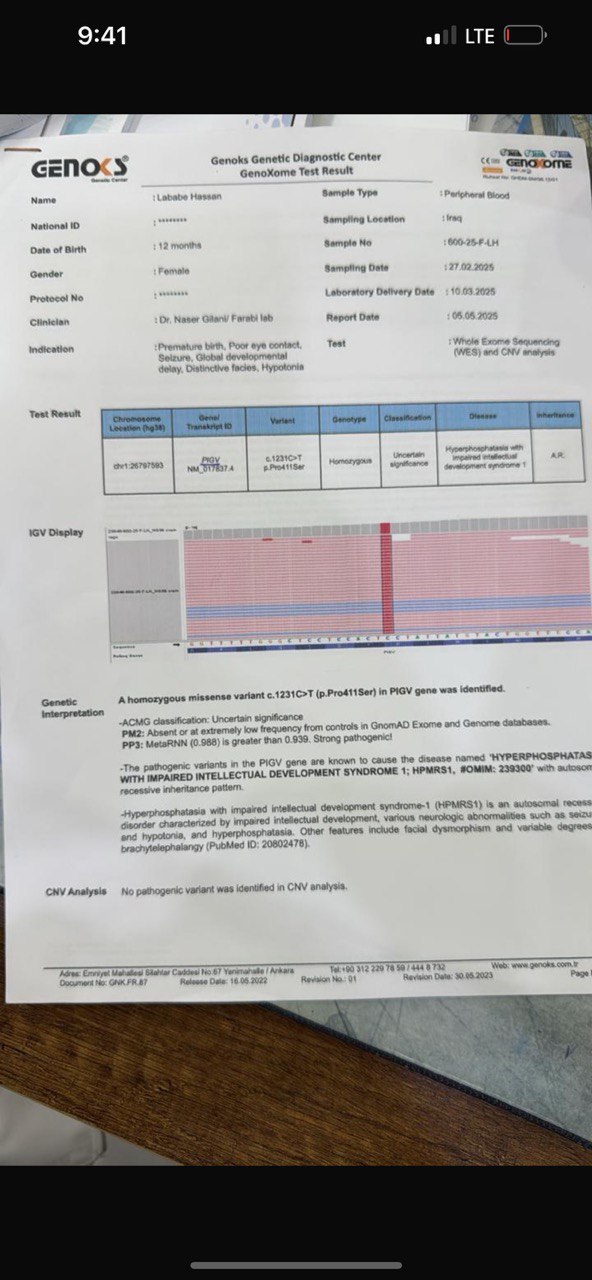
Figure 7: Whole-Exome Sequencing (WES) result – Genox Genetic Diagnostic Center Genetic report confirming a homozygous missense variant c.1231C>T (p.Pro411Ser) in the PIGV gene, consistent with the diagnosis of HPMRS-1
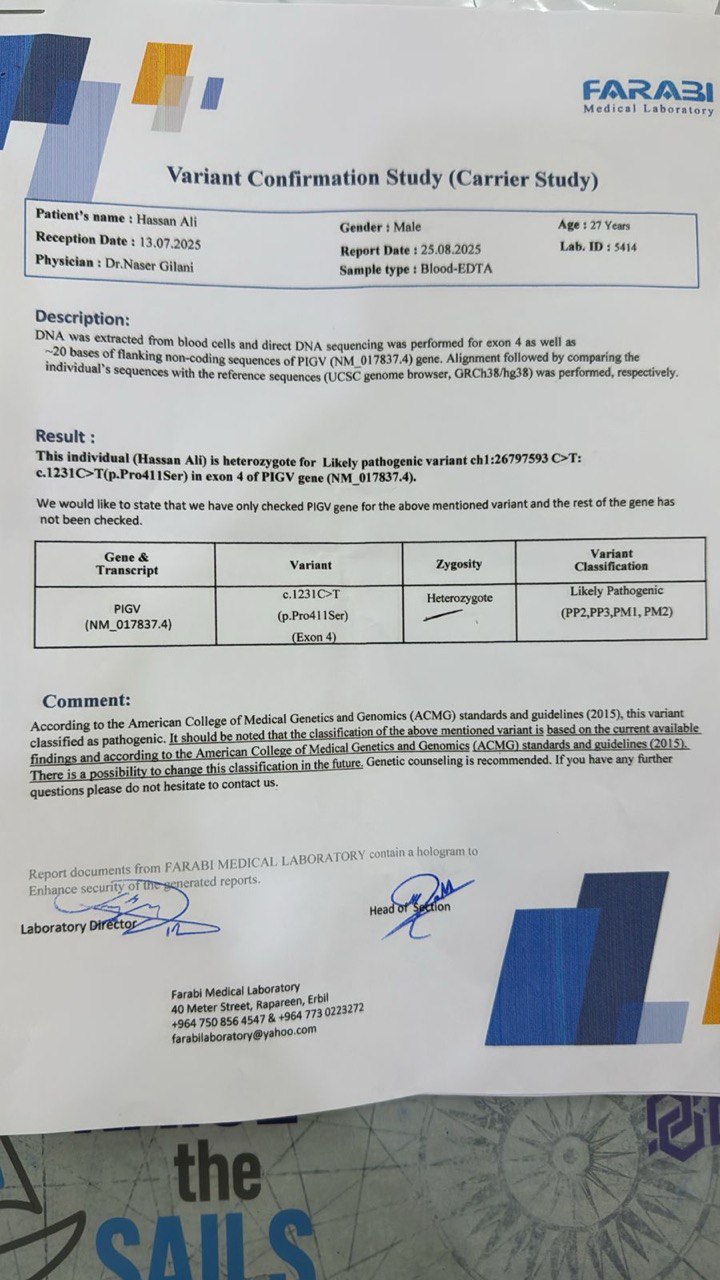
Figure 8: Carrier testing report of the patient’s father – Farabi Medical Laboratory Variant confirmation study showing heterozygous carrier status for the PIGV c.1231C>T (p.Pro411Ser) variant, supporting an autosomal recessive inheritance pattern
Figure 10: Facial features of the patient with HPMRS (Mabry syndrome) Frontal photograph showing coarse facial appearance with a broad forehead, thick eyebrows, long eyelashes, full cheeks, and a flat nasal bridge, typical of PIGV-related HPMRS.
DISCUSSION
This case expands the global literature on HPMRS, representing the first genetically confirmed Iraqi patient. The triad of refractory epilepsy, severe developmental delay, and persistent hyperphosphatasemia strongly suggested a GPI-anchor defect [3]. Early genetic confirmation ensures accurate diagnosis, family counseling, and avoidance of unnecessary procedures [8]. Pathophysiology: The elevated ALP is due to defective membrane anchoring of GPI-linked proteins, especially ALP, causing increased enzyme shedding into circulation [9]. PIGV, the gene mutated in HPMRS-1, encodes mannosyltransferase catalyzing the fifth step of GPI-anchor synthesis [4,9]. Similar biochemical mechanisms are shared by PIGO and PGAP2/3 mutations [5,6].
Clinical Correlation
Our patient’s manifestations—severe hypotonia, seizures, craniofacial dysmorphism, cortical blindness, and skeletal deformities—are consistent with previously reported PIGV-related cases [4,6,10]. Congenital heart disease (ASD) has occasionally been described in this subtype [7,11].
Differential Diagnosis
CHIME syndrome (OMIM 280000), caused by PIGL mutations, shares intellectual disability, seizures, and facial anomalies but includes coloboma, ichthyosis, and ear malformations, which were absent in our case [12,13].
Benign transient hyperphosphatasemia shows isolated ALP elevation without neurological involvement [14].
Figure 11: Skeletal deformities of the upper and lower limbs
Photographs showing short, broad hands with clenched fingers (upper image) and bilateral clubfoot deformities (lower image), consistent with skeletal abnormalities described in HPMRS.
Juvenile Paget disease presents with bone deformities and hearing loss but normal intellect [15].
Therapeutic considerations: While no curative treatment exists, supportive measures such as physiotherapy, nutritional management, and seizure optimization remain essential [8,10]. Some patients benefit from pyridoxine supplementation, reflecting ALP’s role in vitamin B6 metabolism [16]. Our patient, however, showed limited response.
Epidemiologic Significance:
HPMRS accounts for less than 0.5% of developmental disorders [3], yet early recognition using elevated ALP as a screening clue followed by exome sequencing can establish diagnosis, especially in low-resource settings [17]. Reporting such cases enriches global understanding of genotype-phenotype correlations and may promote earlier detection across the Middle East.
CONCLUSION
This is the first genetically confirmed case of HPMRS (PIGV mutation) from Iraq. Persistent hyperphosphatasemia with developmental delay and seizures should prompt consideration of GPI-anchor biosynthesis defects. Early diagnosis is vital for genetic counseling and appropriate multidisciplinary care.
REFERENCE
Mabry, C.C., et al. “Familial Hyperphosphatasia with Mental Retardation, Seizures, and Neurologic Deficit.” Journal of Pediatrics, vol. 77, no. 1, 1970, pp. 74–85.
Krawitz, P.M., et al. “PIGV Mutations Cause Hyperphosphatasia with Mental Retardation Syndrome Type 1 (HPMRS1).” Nature Genetics, vol. 42, no. 7, 2010, pp. 555–559.
Knaus, A., et al. “Characterization of Glycosylphosphatidylinositol Anchor Biosynthesis Defects.” Journal of Inherited Metabolic Disease, vol. 41, no. 3, 2018, pp. 495–505.
Nguyen, T.T.M., et al. “PIGV Gene Mutations and Clinical Spectrum of HPMRS.” Clinical Genetics, vol. 95, no. 6, 2019, pp. 716–725.
Fujita, M., et al. “PIGO Mutations in Hyperphosphatasia with Mental Retardation Syndrome Type 2.” American Journal of Human Genetics, vol. 91, no. 1, 2012, pp. 146–151.
Hansen, L., et al. “PGAP2 and PGAP3 Mutations in GPI-Anchor Deficiencies.” Human Mutation, vol. 34, no. 1, 2013, pp. 146–152.
Martin, H.C., et al. “Clinical Spectrum and Genetics of Congenital GPI-Anchor Deficiencies.” Genetics in Medicine, vol. 19, no. 9, 2017, pp. 986–994.
Brady, P.D., et al. “Diagnostic Utility of Whole-Exome Sequencing in Neurodevelopmental Disorders.” Clinical Genetics, vol. 99, no. 1, 2021, pp. 90–100.
Hong, Y., et al. “Biosynthesis of GPI Anchors and Their Defects.” FEBS Letters, vol. 594, no. 1, 2020, pp. 3–22.
Ranza, E., et al. “PIGV-Related Mabry Syndrome: Expanded Phenotype and Management.” European Journal of Medical Genetics, vol. 64, no. 9, 2021, article 104297.
Ng, B.G., and H.H. Freeze. “Perspectives on Congenital Disorders of Glycosylation and GPI-Anchor Biosynthesis.” Glycobiology, vol. 28, no. 9, 2018, pp. 623–640.
Ng, B.G., et al. “Mutations in PIGL Cause CHIME Syndrome.” American Journal of Human Genetics, vol. 90, no. 4, 2012, pp. 685–688.
Wang, H., et al. “Phenotypic Overlap between CHIME and HPMRS Syndromes Due to PIGL Variants.” Molecular Genetics and Metabolism Reports, vol. 32, 2022, article 100923.
Kraut, J.R., et al. “Benign Transient Hyperphosphatasemia of Infancy and Early Childhood.” Journal of Pediatrics, vol. 106, no. 5, 1985, pp. 718–722.
Whyte, M.P., et al. “Juvenile Paget’s Disease: Molecular and Clinical Findings.” Bone, vol. 59, 2014, pp. 143–153.
Murakami, Y., et al. “Pyridoxine-Responsive Seizures and GPI Anchor Defects.” Brain and Development, vol. 40, no. 3, 2018, pp. 226–233.
Richards, S., et al. “Standards and Guidelines for the Interpretation of Sequence Variants.” Genetics in Medicine, vol. 17, no. 5, 2015, pp. 405–424.
License
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
All papers should be submitted electronically. All submitted manuscripts must be original work that is not under submission at another journal or under consideration for publication in another form, such as a monograph or chapter of a book. Authors of submitted papers are obligated not to submit their paper for publication elsewhere until an editorial decision is rendered on their submission. Further, authors of accepted papers are prohibited from publishing the results in other publications that appear before the paper is published in the Journal unless they receive approval for doing so from the Editor-In-Chief.
Himalayan Journal of Community Medicine and Public Health open access articles are licensed under a Creative Commons Attribution-Share A like 4.0 International License. This license lets the audience to give appropriate credit, provide a link to the license, and indicate if changes were made and if they remix, transform, or build upon the material, they must distribute contributions under the same license as the original.
Recommended Articles
Research Article
Study of fish quality in Hor Abu Zark 2021-2022
Basim Turki Wthiaj Alyousif,
Jameelah Al-Yousef
Published: 24/01/2024
Download PDF
Cite
x
APA
Wthiaj Alyousif, B. T. & Al-Yousef, J. (2024). Study of fish quality in Hor Abu Zark 2021-2022. Himalayan Journal of Community Medicine and Public Health, 5(1), 1-6.
MLA
Wthiaj Alyousif, Basim Turki and Jameelah Al-Yousef. "Study of fish quality in Hor Abu Zark 2021-2022." Himalayan Journal of Community Medicine and Public Health 5.1 (2024): 1-6.
Chicago
Wthiaj Alyousif, Basim Turki and Jameelah Al-Yousef. "Study of fish quality in Hor Abu Zark 2021-2022." Himalayan Journal of Community Medicine and Public Health 5, no. 1 (2024): 1-6.
Harvard
Wthiaj Alyousif, B. T. and Al-Yousef, J. (2024) 'Study of fish quality in Hor Abu Zark 2021-2022' Himalayan Journal of Community Medicine and Public Health 5(1), pp. 1-6.
Vancouver
Wthiaj Alyousif BT, Al-Yousef J. Study of fish quality in Hor Abu Zark 2021-2022. Himalayan Journal of Community Medicine and Public Health. 2024 Jan;5(1):1-6.
Download PDF
Research Article
Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang
Anang Kriswantoro,
...
Swasty
Published: 20/01/2026
Download PDF
Cite
x
APA
Kriswantoro, A., Martiningsih, W. R., Novitasari, A. & None, S. (2026). Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang. Himalayan Journal of Community Medicine and Public Health, 7(1), 1-5.
MLA
Kriswantoro, Anang, et al. "Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang." Himalayan Journal of Community Medicine and Public Health 7.1 (2026): 1-5.
Chicago
Kriswantoro, Anang, Wahju R. Martiningsih, Andra Novitasari and Swasty . "Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang." Himalayan Journal of Community Medicine and Public Health 7, no. 1 (2026): 1-5.
Harvard
Kriswantoro, A., Martiningsih, W. R., Novitasari, A. and None, S. (2026) 'Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang' Himalayan Journal of Community Medicine and Public Health 7(1), pp. 1-5.
Vancouver
Kriswantoro A, Martiningsih WR, Novitasari A, Swasty S. Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang. Himalayan Journal of Community Medicine and Public Health. 2026 Jan;7(1):1-5.
Download PDF
Research Article
Otolaryngologic Perspective on Endoscopic Sellar Reconstruction after Transsphenoidal Pituitary Surgery
Sattar Jaber Abed,
...
Asaad Mezher Hussain
Published: 31/12/2025
Download PDF
Cite
x
APA
Jaber Abed, S., Khazaal Hashim, M. & Mezher Hussain, A. (2025). Otolaryngologic Perspective on Endoscopic Sellar Reconstruction after Transsphenoidal Pituitary Surgery. Himalayan Journal of Community Medicine and Public Health, 6(2), 1-5.
MLA
Jaber Abed, Sattar, Mohammed Khazaal Hashim and Asaad Mezher Hussain. "Otolaryngologic Perspective on Endoscopic Sellar Reconstruction after Transsphenoidal Pituitary Surgery." Himalayan Journal of Community Medicine and Public Health 6.2 (2025): 1-5.
Chicago
Jaber Abed, Sattar, Mohammed Khazaal Hashim and Asaad Mezher Hussain. "Otolaryngologic Perspective on Endoscopic Sellar Reconstruction after Transsphenoidal Pituitary Surgery." Himalayan Journal of Community Medicine and Public Health 6, no. 2 (2025): 1-5.
Harvard
Jaber Abed, S., Khazaal Hashim, M. and Mezher Hussain, A. (2025) 'Otolaryngologic Perspective on Endoscopic Sellar Reconstruction after Transsphenoidal Pituitary Surgery' Himalayan Journal of Community Medicine and Public Health 6(2), pp. 1-5.
Vancouver
Jaber Abed S, Khazaal Hashim M, Mezher Hussain A. Otolaryngologic Perspective on Endoscopic Sellar Reconstruction after Transsphenoidal Pituitary Surgery. Himalayan Journal of Community Medicine and Public Health. 2025 Jul;6(2):1-5.
Download PDF
Research Article
Serum Leptin Levels in Early Pregnancy and Their Association with Unexplained Recurrent Pregnancy Loss: A Prospective Case–Control Study
Wasan Salih Mohammed,
Sali Asif Younus
Published: 05/01/2026
Download PDF
Cite
x
APA
Mohammed, W. S. & Younus, S. A. (2026). Serum Leptin Levels in Early Pregnancy and Their Association with Unexplained Recurrent Pregnancy Loss: A Prospective Case–Control Study. Himalayan Journal of Community Medicine and Public Health, 7(1), 1-5.
MLA
Mohammed, Wasan S. and Sali A. Younus. "Serum Leptin Levels in Early Pregnancy and Their Association with Unexplained Recurrent Pregnancy Loss: A Prospective Case–Control Study." Himalayan Journal of Community Medicine and Public Health 7.1 (2026): 1-5.
Chicago
Mohammed, Wasan S. and Sali A. Younus. "Serum Leptin Levels in Early Pregnancy and Their Association with Unexplained Recurrent Pregnancy Loss: A Prospective Case–Control Study." Himalayan Journal of Community Medicine and Public Health 7, no. 1 (2026): 1-5.
Harvard
Mohammed, W. S. and Younus, S. A. (2026) 'Serum Leptin Levels in Early Pregnancy and Their Association with Unexplained Recurrent Pregnancy Loss: A Prospective Case–Control Study' Himalayan Journal of Community Medicine and Public Health 7(1), pp. 1-5.
Vancouver
Mohammed WS, Younus SA. Serum Leptin Levels in Early Pregnancy and Their Association with Unexplained Recurrent Pregnancy Loss: A Prospective Case–Control Study. Himalayan Journal of Community Medicine and Public Health. 2026 Jan;7(1):1-5.
Laylani, A. A., Abdulzahra, I. K. & Habeed, E. N. (2025). Hyperphosphatasia with Impaired Intellectual Development Syndrome (Mabry Syndrome): First Genetically Confirmed Case from Iraq. Himalayan Journal of Community Medicine and Public Health, 6(2), 1-5.
MLA
Laylani, Amal Adnan, Ihsan Kareem Abdulzahra and Esraa Najat Habeed. "Hyperphosphatasia with Impaired Intellectual Development Syndrome (Mabry Syndrome): First Genetically Confirmed Case from Iraq." Himalayan Journal of Community Medicine and Public Health 6.2 (2025): 1-5.
Chicago
Laylani, Amal Adnan, Ihsan Kareem Abdulzahra and Esraa Najat Habeed. "Hyperphosphatasia with Impaired Intellectual Development Syndrome (Mabry Syndrome): First Genetically Confirmed Case from Iraq." Himalayan Journal of Community Medicine and Public Health 6, no. 2 (2025): 1-5.
Harvard
Laylani, A. A., Abdulzahra, I. K. and Habeed, E. N. (2025) 'Hyperphosphatasia with Impaired Intellectual Development Syndrome (Mabry Syndrome): First Genetically Confirmed Case from Iraq' Himalayan Journal of Community Medicine and Public Health 6(2), pp. 1-5.
Vancouver
Laylani AA, Abdulzahra IK, Habeed EN. Hyperphosphatasia with Impaired Intellectual Development Syndrome (Mabry Syndrome): First Genetically Confirmed Case from Iraq. Himalayan Journal of Community Medicine and Public Health. 2025 Jul;6(2):1-5.