Background: Short stature is a common pediatric concern and may represent normal growth variants or underlying pathological conditions. Early identification and accurate etiological classification are essential for appropriate management and prevention of long-term complications. Despite extensive global data, regional epidemiological information remains limited. Objective: To determine the patterns and etiological distribution of short stature among children attending the growth center at Shar Hospital, Sulaimaniyah, Iraq and to assess the relative frequency of underlying causes. Methods: This retrospective descriptive study was conducted at the growth center at Shar Hospital, Sulaimaniyah, Iraq, from March 2014 to January 2015. A total of 326 children aged 2–17 years with short stature (height<−2 SD or below the 3rd percentile) were included. Data were extracted from medical records and included demographic characteristics, family history, clinical findings, laboratory investigations, bone age assessment and hormonal evaluation. Growth hormone deficiency was confirmed using stimulation tests when indicated. Data were analyzed using descriptive statistical methods. Results: The mean age of patients was 9.6 years, with the highest frequency in the 8–11-year age group (32.5%). Females represented 54.3% of cases. Delayed bone age was observed in 63.8% of patients. Idiopathic short stature was the most common cause (49.8%), followed by familial short stature (19%) and growth hormone deficiency (16.3%). Small for gestational age accounted for 7.4%, while hypothyroidism and Turner syndrome each accounted for 1.8%. Other rare causes collectively represented 4%. Most children did not have significant endocrine or systemic pathology. Conclusion: Idiopathic and familial short stature constitute the majority of cases in this population, whereas endocrine causes represent a smaller proportion. Comprehensive evaluation including detailed history, anthropometric assessment, bone age analysis and targeted laboratory testing is essential for accurate diagnosis. Regional data such as these are valuable for optimizing diagnostic strategies and healthcare planning.
Keywords
Short Stature
Idiopathic Short Stature
Growth Hormone Deficiency
Familial Short Stature
Bone Age
Pediatric Endocrinology
Etiological Profile
Growth Disorders
INTRODUCTION
Growth is a fundamental biological process of childhood and adolescence and reflects the dynamic interaction of genetic, nutritional, hormonal and environmental influences. Linear growth, in particular, is widely recognized as one of the most sensitive indicators of a child’s overall health status and well-being [1,2]. Impaired growth may represent the earliest clinical manifestation of systemic, nutritional, endocrine, or genetic disorders, underscoring the importance of careful monitoring in pediatric practice [3]. On a population level, secular trends in height and growth velocity provide valuable insight into socioeconomic conditions, nutritional adequacy and public health development [4]. The American Academy of Pediatrics (AAP) recommends routine anthropometric assessment throughout childhood, including serial measurement of height and weight plotted on standardized growth charts [5]. Early identification of abnormal growth patterns allows timely evaluation and intervention, thereby minimizing long-term physical and psychosocial consequences [6]. Physiologically, growth velocity decreases after infancy, remains relatively stable during mid-childhood and accelerates during puberty. In girls, the pubertal growth spurt usually begins at 10–11 years, whereas in boys it typically occurs at 12–14 years [7,8]. Deviation from these expected patterns warrants further investigation. Short stature is commonly defined as height below −2 Standard Deviations (SD) for age and sex, corresponding approximately to the third percentile on standardized growth charts [9]. Additional concerning features include reduced growth velocity (less than 4–5 cm per year after the age of five) and downward crossing of two major percentile lines [10]. Differentiating between normal growth variants and pathological causes is crucial. Normal variants include Familial Short Stature (FSS) and Constitutional Growth Delay (CGD), while pathological short stature may result from endocrine, systemic, chromosomal, skeletal, or psychosocial disorders [11]. Familial short stature is characterized by a height consistent with parental genetic potential, normal growth velocity and normal bone age [12,13]. In contrast, constitutional growth delay involves delayed skeletal maturation and delayed puberty, often with a positive family history of delayed growth or “late bloomers,” with eventual attainment of normal adult height [14]. Pathological causes, although less frequent, are clinically significant. Endocrine disorders-particularly Growth Hormone Deficiency (GHD), hypothyroidism and Cushing syndrome-represent important etiologies [15]. GHD may be congenital, acquired, genetic, or idiopathic, with an estimated incidence of 1 in 4,000 to 10,000 live births [16]. Diagnosis relies on assessment of serum insulin-like growth factor-1 (IGF-1), IGF binding protein-3 and growth hormone stimulation testing [17].
The Growth Hormone–Insulin-like Growth Factor (GH–IGF) axis plays a central role in postnatal linear growth. GH, secreted in a pulsatile manner from the anterior pituitary, stimulates hepatic production of IGF-1, which mediates chondrocyte proliferation and longitudinal bone growth [18]. Disruption at any level of this axis-including mutations affecting GH secretion, GH receptors, or downstream signaling pathways-may result in impaired linear growth [19,20]. Advances in molecular endocrinology have refined the understanding of children previously labeled as having Idiopathic Short Stature (ISS), highlighting the heterogeneity of this diagnosis.
Chromosomal abnormalities, particularly Turner syndrome, must be considered in any short girl regardless of phenotypic presentation. Chronic systemic illnesses, intrauterine growth restriction, malnutrition and inflammatory disorders can also adversely affect growth. Accurate assessment requires serial measurements, bone age evaluation and correlation with mid-parental height expectations. Despite extensive international data, regional patterns of short stature vary according to genetic background, nutritional status and healthcare accessibility. In the Kurdistan region, including Kirkuk city, epidemiological data on the etiological spectrum of short stature remain limited. Therefore, this study aimed to determine the pattern of short stature among children attending the growth center at Shar Hospital, Sulaimaniyah, Iraq and to assess the frequency of underlying etiological factors in this population.
MATERIALS AND METHODS
This retrospective descriptive study was conducted at the growth center at Shar Hospital, Sulaimaniyah, Iraq, during the period from March 2014 to January 2015. The study included 326 children diagnosed with short stature who attended the growth clinic within the specified duration. Patients’ files were randomly selected from medical records and all relevant clinical, laboratory and radiological data were extracted and analyzed. Only children aged between 2 and 17 years who met the diagnostic criteria for short stature were included. Short stature was defined as height below −2 standard deviations (−2 SD) for age and sex or height less than the 3rd percentile according to standardized growth charts. Patients younger than 2 years of age and files with incomplete or insufficient documentation were excluded from the study.
All patient records were reviewed using a structured data collection sheet. The information obtained included age at presentation, sex, residency (urban or rural), age at initiation of growth hormone therapy if applicable, family history of short stature, family history of Constitutional Growth Delay (CGD), history of precocious puberty and presence of chronic systemic illnesses. Each patient had undergone comprehensive clinical evaluation at the time of presentation. Anthropometric measurements included standing height measured using a wall-mounted stadiometer, height percentile plotted on standardized growth charts and growth velocity prior to treatment when available. Body weight and weight percentile were also recorded. Parental heights were documented and Mid-Parental Height (MPH) was calculated using standard formulas to assess genetic growth potential. The child’s projected height was compared with the calculated mid-parental height percentile.
A thorough general and systemic examination had been performed for all patients to identify features suggestive of endocrine, genetic, skeletal, or systemic causes of short stature. Particular attention was given to dysmorphic features, disproportionate body segments, signs of chronic disease and clinical manifestations of hypothyroidism or hypercortisolism. In male patients, pubertal status was assessed by measuring testicular size using orchidometry.
Laboratory investigations were performed according to clinical indications to determine the underlying etiology. Hematological tests included Complete Blood Picture (CBP) and erythrocyte sedimentation rate (ESR). Biochemical tests included blood glucose level, blood urea and serum electrolytes, serum ferritin, serum anti-tissue transglutaminase antibodies and serum vitamin D level. Hormonal evaluation included measurement of serum insulin-like growth factor-1 (IGF-1), which was expressed in ng/mL and plotted on standardized age- and sex-adjusted reference charts. IGF-1 values were categorized relative to standard deviation ranges (Mean, −1 SD, −2 SD, −3 SD, +1 SD, +2 SD, +3 SD). Basal Growth Hormone (GH) levels were measured and GH stimulation tests were performed using exercise, clonidine, or glucagon protocols when indicated. Thyroid function tests, including serum TSH and free T4, were assessed to exclude hypothyroidism. In selected cases, serum LH, FSH, cortisol and estradiol levels were measured based on clinical suspicion.
Radiological assessment included determination of bone age using plain radiography of the left hand and wrist, which was compared with standard reference values appropriate for chronological age. Pelvic ultrasound was performed in female patients suspected of Turner syndrome. Magnetic Resonance Imaging (MRI) of the brain was conducted in patients diagnosed with growth hormone deficiency to evaluate hypothalamic-pituitary abnormalities. Additional investigations were carried out in selected patients as clinically indicated, including upper gastrointestinal endoscopy in cases with positive anti-tissue transglutaminase antibodies to confirm celiac disease and genetic studies when chromosomal or molecular abnormalities were suspected.
After complete clinical, laboratory and radiological evaluation, patients were categorized into diagnostic groups. Idiopathic Short Stature (ISS) included patients with normal general examination, normal IGF-1 levels, growth velocity below the 25th percentile and bone age delayed by 1–2 years relative to chronological age.
Familial (genetic) short stature included patients with normal examination findings, normal growth velocity, normal bone age, normal IGF-1 levels and height consistent with calculated mid-parental height. Growth Hormone Deficiency (GHD) was diagnosed in patients who had serum IGF-1 below −2 SD for age and sex, reduced growth velocity (10–25th percentile), delayed bone age and an abnormal GH stimulation test (Figure 1).
Figure 1: Age-Specific Height Velocity Curves in Girls and Boys: Colored Percentile Growth Patterns with Pubertal Peak Velocity Zones
All collected data were entered and analyzed using appropriate statistical software. Continuous variables were expressed as mean ±Standard Deviation (SD) and categorical variables were presented as frequencies and percentages. Patient confidentiality was maintained throughout the study and no identifying information was disclosed. This methodological approach enabled systematic evaluation of the etiological patterns of short stature among children attending the Pediatric Growth Clinic at Children Hospital in Kirkuk city during the study period (Figure 2).
Figure 2: Age- and Sex-Specific Reference Curves for Serum IGF-1 Levels (Mean and Standard Deviation Ranges) in Children and Adolescents
RESULTS
The ages of the patients included in this study ranged from 2 to 17 years, with a mean age of 9.6 years. The most represented age group was 8–11 years, accounting for 106 patients (32.5%), followed by the 11–14 years’ group with 91 patients (27.9%). The detailed age distribution is presented in Table 1.
Table 1: Age Distribution of the Patients
Age Group (years)
No. of Patients
Percentage
2–5
34
10.4
>5–8
57
17.5
>8–11
106
32.5
>11–14
91
27.9
>14–17
38
11.7
Total
326
100
Out of the 326 patients enrolled in this study, 177 (54.3%) were females and 149 (45.7%) were males. This indicates a slight female predominance in the study population (Table 2).
Table 2: Sex Distribution of the Patients
Sex
No. of Patients
Percentage
Female
177
54.3
Male
149
45.7
Total
326
100
Regarding place of residence, 174 patients (53.4%) were from urban areas, while 152 patients (46.6%) were from rural areas. The distribution is shown in Table 3.
Table 3: Residence Distribution of the Patients
Residence
No. of Patients
Percentage
Urban
174
53.4
Rural
152
46.6
Total
326
100
Most patients had no significant family history. However, positive family history was reported in 84 cases (27.5%) distributed as follows: Familial (genetic) short stature: 44 cases (13.5%) Constitutional Growth Delay (CGD): 37 cases (11.3%) Precocious puberty: 3 cases (0.9%) These findings are summarized in Table 4.
Table 4: Family History among the Studied Patients
Family History Condition
No. of Patients
(%)
Familial (genetic) short stature
44
13.5
Constitutional Growth Delay (CGD)
37
11.3
Precocious puberty
3
0.9
Total
84
27.5
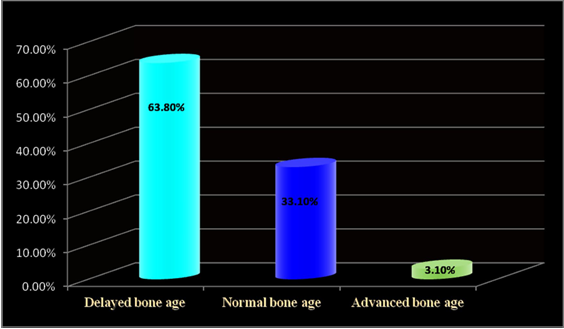
Bone age was evaluated in all patients using plain wrist radiography. The findings revealed: Delayed bone age in 208 patients (63.8%), Normal bone age in 108 patients (33.1%), Advanced bone age in 10 patients (3.1%), These results indicate that delayed bone age was the most common radiological finding among the studied population (Figure 3).
Figure 3: Bone Age Among Our Cases
Out of the 209 cases referred for thyroid function testing, hypothyroidism was identified in only six patients (2.87%), characterized by low T4 and elevated TSH levels. The remaining 203 cases (97.13%) demonstrated normal thyroid hormone profiles. No cases of hyperthyroidism were detected among the studied patients.
Regarding serum vitamin D assessment, 274 patients (84%) had normal serum vitamin D levels (8–80 ng/ml), whereas 52 patients (16%) exhibited low levels. No cases of elevated serum vitamin D were documented.
Serum ferritin levels were within the normal range for age and sex in 284 patients (87.1%). Low ferritin levels were observed in 39 patients (12%), while elevated ferritin levels were found in three patients (0.9%), two of whom were diagnosed with thalassemia.
Concerning serum anti-transglutaminase antibodies, only one case (0.3%) showed elevated levels. Subsequent diagnostic evaluation in this patient confirmed underlying celiac disease.
Assessment of growth hormone (GH) revealed that 314 patients (96.3%) had normal basal GH levels (normal range: 0–20 ng/ml), while 12 patients (3.7%) exhibited decreased basal levels. Growth hormone provocation testing, performed using exercise, glucagon, or clonidine stimulation, demonstrated classical GH deficiency (serum level <5 ng/ml) in 35 patients (9.3%) and partial GH deficiency (5–10 ng/ml) in 18 patients (18%). The remaining 273 patients (70.2%) were considered not to have GH deficiency based on the provocation test results.
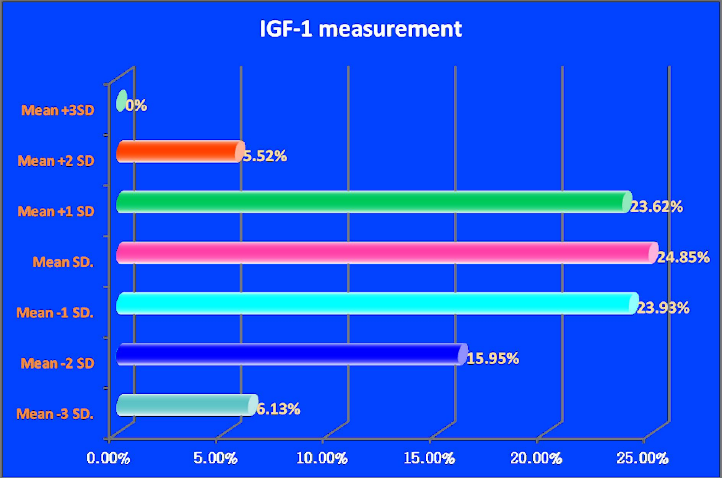
For insulin-like growth factor-1 (IGF-1) evaluation, serum IGF-1 levels were measured in ng/ml and plotted on standardized charts adjusted for age and sex. The values were categorized according to their deviation from the mean as follows: mean, mean −1 SD, mean −2 SD, mean −3 SD, mean +1 SD, mean +2 SD and mean +3 SD. The distribution of IGF-1 levels is illustrated in Figure 4.
Figure 4: Results of GF-1
These additional investigations were selectively performed in patients when there was strong clinical suspicion of specific underlying etiologies or associated disorders. The investigations carried out and their significant findings are summarized in Table 5.
Table 5: Specific Investigations Performed in Selected Cases
Specific Investigation
Clinical Purpose / Finding
MRI of brain
One case was diagnosed with a brain cyst.
Chromosomal analysis (Karyotyping)
Six cases were diagnosed with Turner’s syndrome.
Upper gastrointestinal endoscopy
Performed in patients with positive anti-transglutaminase antibodies; confirmed celiac disease in one case.
Serum LH, FSH, cortisol and estradiol assays
Used for evaluation of pubertal disorders; assisted in diagnosing precocious puberty in six cases and supported assessment in suspected Turner’s syndrome.
After comprehensive clinical, laboratory and radiological evaluation of the 326 patients included in this study, the underlying causes of short stature were identified as shown in Table 6.
Table 6: Causes of Short Stature among the Studied Patients (n = 326)
Causes
Total Cases (No.)
%
Female (No.)
%
Male (No.)
%
Idiopathic SS
162
49.8
74
22.8
88
27.0
Familial (Genetic) SS
62
19.0
35
10.7
27
8.3
GH Deficiency
53
16.3
38
11.7
15
4.6
Small for Gestational Age
24
7.4
16
4.9
8
2.5
Hypothyroidism
6
1.8
3
0.9
3
0.9
Turner’s Syndrome
6
1.8
6
1.8
–
–
Other rare causes*
13
4.0
5
1.5
8
2.5
Total
326
100
177
54.3
149
45.7
*Other rare causes include Russell–Silver syndrome, chronic renal disease, Noonan syndrome, Prader–Willi syndrome, hypopituitarism, celiac disease, mucopolysaccharidosis and hypochondroplasia
Idiopathic short stature (ISS) was the most common cause, accounting for 162 cases (49.8%), followed by familial (genetic) short stature in 62 cases (19%). Growth Hormone (GH) deficiency was diagnosed in 53 cases (16.3%). Other causes included Small for Gestational Age (SGA), hypothyroidism, Turner’s syndrome and a limited number of syndromic or systemic disorders. Prader–Willi syndrome, hypopituitarism, celiac disease, mucopolysaccharidosis and hypochondroplasia.
DISCUSSION
The present study evaluated the pattern and etiological profile of short stature among children attending the growth center at Shar Hospital, Sulaimaniyah, Iraq through comprehensive clinical, biochemical and radiological assessment. The age of the patients ranged from 2 to 17 years, with a mean age of 9.6 years and the largest proportion of cases occurred in the 8–11-year age group (32.5%), followed by 11–14 years (27.9%) and 5–8 years (17.5%), suggesting that growth delay becomes more noticeable and prompts medical consultation during mid-childhood. These findings are comparable to those reported by Shazia et al. [10] and Marjolein et al. [14], who documented similar mean ages at presentation. Females constituted 54.3% of cases, showing slight female predominance, in agreement with Shazia et al. [10] and Nada A. Ahmed et al. [13], although other studies such as Nasir Al-Jurayyan et al. [9] and El Mouzan et al. [11] reported male predominance, possibly reflecting regional and sociocultural differences in referral patterns. More than half of the patients (53.4%) were from urban areas, likely due to better awareness and access to healthcare facilities. A positive family history of short stature or constitutional growth delay was identified in 24.8% of cases, closely matching the 25% reported by Shazia et al. [10], highlighting the importance of genetic and constitutional factors. Delayed bone age was observed in 63.8% of patients, higher than that reported by Nada A. Ahmed et al. [13], which may reflect differences in patient selection or referral bias. IGF-1 levels clustered mainly around the mean and −1 SD, supporting its role as a useful adjunct in screening for GH deficiency, as noted by Juul et al. [21], although it remains a supportive rather than definitive diagnostic marker. Regarding etiology, Idiopathic Short Stature (ISS) was the most common cause (49.8%), followed by familial short stature (19%) and growth hormone deficiency (16.3%), confirming that shortest children do not have serious endocrine or systemic pathology. These findings are consistent with reports by Shazia et al. [10] and Moayeri and Aghighi [20], although Nasir Al-Jurayyan et al. [9] found genetic short stature to be predominant. The prevalence of GH deficiency in our study falls within the range reported internationally (7.4–23.4%) [17-19]. Turner syndrome and hypothyroidism each accounted for 1.8% of cases. Idiopathic short stature was the leading cause in both sexes, followed by GH deficiency in females and familial short stature in males, differing slightly from some regional reports. In conclusion, idiopathic and familial short stature constitute the majority of cases and endocrine disorders account for a smaller proportion. Proper evaluation should include detailed history, physical examination, bone age assessment and targeted laboratory investigations; however, many children will remain classified as idiopathic despite thorough assessment. The study is limited by its single-center design, potential confounding environmental factors and limited access to advanced genetic testing and future multicenter studies with molecular diagnostic support are recommended.
REFERENCES
Lifshitz, F. Pediatric Endocrinology: Growth, Adrenal, Sexual, Thyroid, Calcium and Fluid Balance Disorders. 5th ed., vol. 2, Informa Healthcare, 2007, pp. 1–9, 40–51.
Polin, R.A. and M.F. Ditmar. Pediatric Secrets. 5th ed., Mosby Elsevier, 2011, pp. 211–215.
Hay, W.W. et al. Current Diagnosis and Treatment: Pediatrics. 20th ed., McGraw-Hill, 2010, ch. 32.
Burg, F.D. et al. Current Pediatric Therapy. 18th ed., Saunders, 2006, sec. 14.
Custer, J.W. and R.E. Rau. The Harriet Lane Handbook. 18th ed., Mosby Elsevier, 2008, ch. 10.
Bernstein, S. et al. Pediatrics: Endocrinology. Part 1, 2006, pp. 9–28.
Sondheimer, J.M. Current Essentials Pediatrics. 1st ed., McGraw-Hill, 2008, pp. 173–174.
Kliegman, R.M. et al. Nelson Textbook of Pediatrics. 19th ed., Elsevier, 2011, ch. 13, pp. 39–43.
Al-Jurayyan, N.A. et al. “Short stature in children: pattern and frequency in a pediatric clinic, Riyadh, Saudi Arabia.” Sudan Journal of Paediatrics, vol. 12, 2012, pp. 79–83.
Lashari, S.K. et al. “Frequency of etiological factors in short statured patients presenting at an endocrine clinic of a tertiary care hospital.” 2007, pp. 1–8.
El Mouzan, M.I. et al. “Prevalence of short stature in saudi children and adolescents.” Annals of Saudi Medicine, vol. 31, no. 5, 2011, pp. 498–501.
Bettendorf, M. et al. “Metacarpal index in short stature before and during growth hormone treatment.” Archives of Disease in Childhood, vol. 79, 1998, pp. 165–168.
Ahmed, N.A. and K.A. Sahar. “Short stature in children: pathological causes.” Tikrit Medical Journal, vol. 14, no. 1, 2008, pp. 211–214.
Bonthuis, M. et al. “Application of body mass index according to height-age in short and tall children.” 2013, pp. 1–6.
Hashemi, J. et al. “Prevalence of celiac disease in Iranian children with idiopathic short stature.” World Journal of Gastroenterology, vol. 14, no. 48, 2008, pp. 7376–7380.
Nogueira de Almeida, C.A. et al. “Growth and hematological studies on brazilian children of low socioeconomic level.”
Bhadda, S.K. et al. “Etiological profile of short stature.” Indian Journal of Pediatrics, vol. 70, 2003, pp. 545–547.
Zargar, A.H. et al. “An etiological profile of short stature in the Indian subcontinent.” Pediatric Child Health, vol. 34, no. 6, 1998, pp. 571–576.
Rabbani, M.W. et al. “Causes of short stature identified at a tertiary care hospital in Multan Pakistan.” Pakistan Journal of Medical Sciences, vol. 29, 2013, pp. 53–57.
Moayeri, H. and Y. Aghighi. “A prospective study of etiology of short stature in 426 short children and adolescents.” Archives of Iranian Medicine, vol. 7, 2004, pp. 23–27.
Juul, A. and N.E. Skakkebaek. “Prediction of the outcome of growth hormone provocative testing in short children by measurement of serum levels of insulin-like growth factor i and insulin-like growth factor binding protein 3.” Journal of Pediatrics, vol. 130, no. 2, 1997, pp. 197–204.
License
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
All papers should be submitted electronically. All submitted manuscripts must be original work that is not under submission at another journal or under consideration for publication in another form, such as a monograph or chapter of a book. Authors of submitted papers are obligated not to submit their paper for publication elsewhere until an editorial decision is rendered on their submission. Further, authors of accepted papers are prohibited from publishing the results in other publications that appear before the paper is published in the Journal unless they receive approval for doing so from the Editor-In-Chief.
Himalayan Journal of Community Medicine and Public Health open access articles are licensed under a Creative Commons Attribution-Share A like 4.0 International License. This license lets the audience to give appropriate credit, provide a link to the license, and indicate if changes were made and if they remix, transform, or build upon the material, they must distribute contributions under the same license as the original.
Recommended Articles
Research Article
Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang
Anang Kriswantoro,
...
Swasty
Published: 20/01/2026
Download PDF
Cite
x
APA
Kriswantoro, A., Martiningsih, W. R., Novitasari, A. & None, S. (2026). Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang. Himalayan Journal of Community Medicine and Public Health, 7(1), 1-5.
MLA
Kriswantoro, Anang, et al. "Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang." Himalayan Journal of Community Medicine and Public Health 7.1 (2026): 1-5.
Chicago
Kriswantoro, Anang, Wahju R. Martiningsih, Andra Novitasari and Swasty . "Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang." Himalayan Journal of Community Medicine and Public Health 7, no. 1 (2026): 1-5.
Harvard
Kriswantoro, A., Martiningsih, W. R., Novitasari, A. and None, S. (2026) 'Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang' Himalayan Journal of Community Medicine and Public Health 7(1), pp. 1-5.
Vancouver
Kriswantoro A, Martiningsih WR, Novitasari A, Swasty S. Etiological Analysis of the Incident of Computer Vision Syndrome (Cvs) in Students of the Faculty of Medical Muhammadiyah University Semarang. Himalayan Journal of Community Medicine and Public Health. 2026 Jan;7(1):1-5.
Download PDF
Research Article
Curves of Concern: Public Awareness of Scoliosis and Its Impact on Childhood Development in Hamirpur
Vishal Dhatwalia,
Swati Chandel
Published: 10/12/2024
Download PDF
Cite
x
APA
Dhatwalia, V. & Chandel, S. (2024). Curves of Concern: Public Awareness of Scoliosis and Its Impact on Childhood Development in Hamirpur. Himalayan Journal of Community Medicine and Public Health, 5(2), 1-6.
MLA
Dhatwalia, Vishal and Swati Chandel. "Curves of Concern: Public Awareness of Scoliosis and Its Impact on Childhood Development in Hamirpur." Himalayan Journal of Community Medicine and Public Health 5.2 (2024): 1-6.
Chicago
Dhatwalia, Vishal and Swati Chandel. "Curves of Concern: Public Awareness of Scoliosis and Its Impact on Childhood Development in Hamirpur." Himalayan Journal of Community Medicine and Public Health 5, no. 2 (2024): 1-6.
Harvard
Dhatwalia, V. and Chandel, S. (2024) 'Curves of Concern: Public Awareness of Scoliosis and Its Impact on Childhood Development in Hamirpur' Himalayan Journal of Community Medicine and Public Health 5(2), pp. 1-6.
Vancouver
Dhatwalia V, Chandel S. Curves of Concern: Public Awareness of Scoliosis and Its Impact on Childhood Development in Hamirpur. Himalayan Journal of Community Medicine and Public Health. 2024 Jul;5(2):1-6.
Download PDF
Research Article
Silent Threats Unveiled: Gauging Public Awareness and Understanding of Blood Clotting Disorders in Ambala, Haryana
Ashmita Joshi,
...
Sanjeev Kumar
Published: 30/07/2024
Download PDF
Cite
x
APA
Joshi, A., kaur, J. & Kumar, S. (2024). Silent Threats Unveiled: Gauging Public Awareness and Understanding of Blood Clotting Disorders in Ambala, Haryana. Himalayan Journal of Community Medicine and Public Health, 5(2), 1-6.
MLA
Joshi, Ashmita, Jasleen kaur and Sanjeev Kumar. "Silent Threats Unveiled: Gauging Public Awareness and Understanding of Blood Clotting Disorders in Ambala, Haryana." Himalayan Journal of Community Medicine and Public Health 5.2 (2024): 1-6.
Chicago
Joshi, Ashmita, Jasleen kaur and Sanjeev Kumar. "Silent Threats Unveiled: Gauging Public Awareness and Understanding of Blood Clotting Disorders in Ambala, Haryana." Himalayan Journal of Community Medicine and Public Health 5, no. 2 (2024): 1-6.
Harvard
Joshi, A., kaur, J. and Kumar, S. (2024) 'Silent Threats Unveiled: Gauging Public Awareness and Understanding of Blood Clotting Disorders in Ambala, Haryana' Himalayan Journal of Community Medicine and Public Health 5(2), pp. 1-6.
Vancouver
Joshi A, kaur J, Kumar S. Silent Threats Unveiled: Gauging Public Awareness and Understanding of Blood Clotting Disorders in Ambala, Haryana. Himalayan Journal of Community Medicine and Public Health. 2024 Jul;5(2):1-6.
Download PDF
Research Article
Knowledge About HBV Infection and Its Association with HBSAG Positivity among Residents of Kaza Sub- Division Of District Lahaul & Spiti In Himachal Pradesh
Brij Sharma,
...
Amit Sachdeva
Published: 30/07/2024
Download PDF
Cite
x
APA
Sharma, B., Gupta, A., Sharma, R., Bodh, V., Chauhan, A., None, D., Sharma, N., Sharma, S. & Sachdeva, A. (2024). Knowledge About HBV Infection and Its Association with HBSAG Positivity among Residents of Kaza Sub- Division Of District Lahaul & Spiti In Himachal Pradesh. Himalayan Journal of Community Medicine and Public Health, 5(2), 1-8.
MLA
Sharma, Brij, et al. "Knowledge About HBV Infection and Its Association with HBSAG Positivity among Residents of Kaza Sub- Division Of District Lahaul & Spiti In Himachal Pradesh." Himalayan Journal of Community Medicine and Public Health 5.2 (2024): 1-8.
Chicago
Sharma, Brij, Anmol Gupta, Rajesh Sharma, Vishal Bodh, Aashish Chauhan, Dipesh , Neetu Sharma, Sidhant Sharma and Amit Sachdeva. "Knowledge About HBV Infection and Its Association with HBSAG Positivity among Residents of Kaza Sub- Division Of District Lahaul & Spiti In Himachal Pradesh." Himalayan Journal of Community Medicine and Public Health 5, no. 2 (2024): 1-8.
Harvard
Sharma, B., Gupta, A., Sharma, R., Bodh, V., Chauhan, A., None, D., Sharma, N., Sharma, S. and Sachdeva, A. (2024) 'Knowledge About HBV Infection and Its Association with HBSAG Positivity among Residents of Kaza Sub- Division Of District Lahaul & Spiti In Himachal Pradesh' Himalayan Journal of Community Medicine and Public Health 5(2), pp. 1-8.
Vancouver
Sharma B, Gupta A, Sharma R, Bodh V, Chauhan A, Dipesh D, Sharma N, Sharma S, Sachdeva A. Knowledge About HBV Infection and Its Association with HBSAG Positivity among Residents of Kaza Sub- Division Of District Lahaul & Spiti In Himachal Pradesh. Himalayan Journal of Community Medicine and Public Health. 2024 Jul;5(2):1-8.
Rasheed, S. R., Abdulqader, M. E. & Jehad, A. H. (2026). Etiological Patterns of Short Stature Among Children Attending the Growth Center at Shar Hospital, Sulaimaniyah, Iraq. Himalayan Journal of Community Medicine and Public Health, 7(1), 1-5.
MLA
Rasheed, Sirwa Raoof, Mustafa Elias Abdulqader and Ahmed Hasan Jehad. "Etiological Patterns of Short Stature Among Children Attending the Growth Center at Shar Hospital, Sulaimaniyah, Iraq." Himalayan Journal of Community Medicine and Public Health 7.1 (2026): 1-5.
Chicago
Rasheed, Sirwa Raoof, Mustafa Elias Abdulqader and Ahmed Hasan Jehad. "Etiological Patterns of Short Stature Among Children Attending the Growth Center at Shar Hospital, Sulaimaniyah, Iraq." Himalayan Journal of Community Medicine and Public Health 7, no. 1 (2026): 1-5.
Harvard
Rasheed, S. R., Abdulqader, M. E. and Jehad, A. H. (2026) 'Etiological Patterns of Short Stature Among Children Attending the Growth Center at Shar Hospital, Sulaimaniyah, Iraq' Himalayan Journal of Community Medicine and Public Health 7(1), pp. 1-5.
Vancouver
Rasheed SR, Abdulqader ME, Jehad AH. Etiological Patterns of Short Stature Among Children Attending the Growth Center at Shar Hospital, Sulaimaniyah, Iraq. Himalayan Journal of Community Medicine and Public Health. 2026 Jan;7(1):1-5.