Anti-Neutrophil Cytoplasm Antibody (ANCA)-Associated Vasculitis (AAV), which includes Microscopic Polyangiitis (MPA), Granulomatosis with Polyangiitis (GPA) (formerly Wegener’s Granulomatosis) and Eosinophilic Granulomatosis with Polyangiitis (EGPA) (formerly Churg-Strauss syndrome), was defined as a group of pauci-immune necrotizing vasculitides [1,2]. Annual incidence rates are 9.8 per million for Wegener’s granulomatosis [3]. An environmental or infectious trigger in a genetically predisposed individual, leading to production of Anti-Neutrophil Cytoplasmic Antibody (ANCA), pathogenic autoantibodies, can lead to this necrotizing disease of small and medium-vessel. The most common symptoms at the time of presentation are ear, nose and throat involvement (70-100%), pulmonary involvement (50-90%) and renal involvement (40-100%) [4]. Central nervous system involvement (7-11%) is rare [5].
Case Presentation
A 45-year-old male presented to the emergency with sudden onset giddiness one day before admission followed by multiple episodes of vomiting associated with headache. He complained of fever and headache on and off since the last 3 months. At presentation, he had pallor, but no icterus, clubbing, cyanosis, edema, lymphadenopathy or no raised jugular venous pulse.
Vitals in the Emergency department were as follows: temperature of 98.5 degrees Fahrenheit, pulse rate of 120 per minute, respiratory rate of 22 breaths per minute, blood pressure of 140/60 mmHg and oxygen saturation of 98% in room air. There was a slight slurring of speech, but the patient was conscious and oriented. There was bilateral air entry in chest, abdomen was soft and non-tender, S1and S2 heart sounds were audible and there was no facial or limb weakness.
Non-contrast Computerized Tomography of the brain on day of admission showed patchy gyral bleed in para sagittal parieto occipital lobe left side with adjoining white matter edema, localised hematoma measuring ~ 32.1 x 18.5 mm in parietal lobe left side with perifocal edema. Thin hyperdense subdural bleed seen along tentorium left side. 15 days later, at follow up, there was marked reduction intra-parenchymal haemorrhage in left parietal region and reduction of Subdural Haemorrhage (SDH) along left fronto-temporo-parietal convexity.
MRI Brain with cerebral angiogram revealed multi focal non-enhancing acute parenchymal hematoma - larger in left parietal and occipital lobe. Thin acute to sub-acute sub dural bleed on the left side with mild patchy diffuse gyral swelling was reported (Figure 1).
Enhancing peripheral dura-based nodule in left parietal region likely to be Granulomatous nodule / Granulomatous angitis was also reported (Figure 2).
Normal course, calibre and branching pattern of vertebro-basillar arteries were obtained in MR angiography. MR venogram was unremarkable. There was no sizeable arterio-venous malformation.
His laboratory results were as follows: activated partial thromboplastin test 45.1 (26 to 40 seconds), prothrombin time 18.7 (11 to 16 seconds), INR 1.48 (0.8-1.1), haemoglobin 7.6 (13-17 g/dl), RBC count 2.86 (4.5 to 5.5 million cells per microliter), packed cell volume 23.1% (40-50%), MCV 80.8 (83-101femtolitre), MCH 26.6 (27-32 picogram), MCHC 32.9 (31.5-34.5 gram/deciliter) RDW 15.4 (11.5-15 %). Platelet and leucocyte count was within reference range. His erythrocyte sedimentation rate was elevated 120 (0-10 millimeter/hour). Peripheral blood smear showed microcytic hypochromic red cells. His iron level was 34.12 (65-175 ug/dl), total iron binding capacity 168.42 (224-428 ug/dl), iron saturation 20.2 (20-50%), ferritin 320(30-400 ng/ml). Stool for occult blood loss tested negative. Based on clinical and laboratory findings anemia was classified as anemia of chronic disease.
His biochemical tests like electrolytes, liver, lipid profiles were within reference range and serology was non-reactive for HIV, Hepatitis B and C.
His urinalysis (UA) showed pH 5, specific gravity 1.020, 4-5 Red Blood Cells (RBC) per High Powered Field (HPF), 1-2 WBCs per HPF, few urine bacteria, urine protein 1+ and no ketones, bilirubin, nitrites, glucose, casts.
Based on history of fever and headache on and off, multiple small acute to subacute intra cerebral haemorrhages and a dura based intracranial granuloma in the setting of anemia of chronic disease we thought of inflammatory or infectious small vessel vasculitis. CT chest shows no pulmonary lesion. Mantoux test was weakly positive (14mm); however, Tuberculosis interferon-gamma release assay was negative. Bacterial endocarditis excluded with normal echocardiogram. Syphilis antibody assay tested negative. Sonography of abdomen shows mild hepatosplenomegaly. Collagen vascular disease excluded with negative rheumatoid factor and Anti-Nuclear Antibody (ANA).
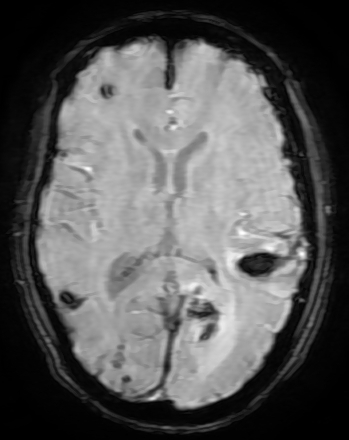
Figure 1: SW MR Image Showing Multiple Acute Haemorrhages
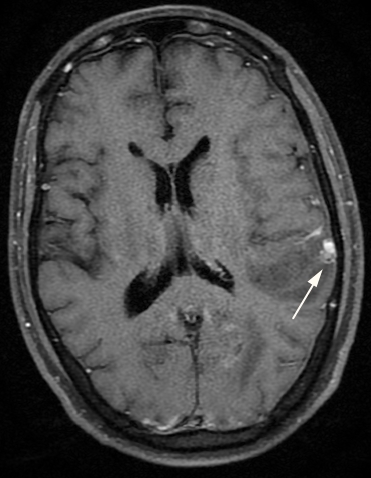
Figure 2: Contrast MR Images Showing Left Parietal Dura Based Enhancing Nodule (Arrow)
Serum PR3 ANCA tested positive with high titer 77.8 IU/ml (negative <5) and serum MPO ANCA was 1.5 (negative <5).
We diagnosed the case as AAV restricted to CNS based on ANCA being positive, MRI brain findings, in the background of constitutional features, anemia and high ESR. Histopathology of the affected organ is gold standard for diagnosis, which was not feasible in our case. Conditions, which can have similar presentation, have been excluded clinically and with the help of ancillary testing. AAV is systemic vasculitis commonly affected organs are lungs, kidneys, heart, ear, nose, throat, orbit skin and peripheral nerves. At this stage, disease seems to be limited to CNS. According to commonly used classification proposed by Watts’ [6], patient remains unclassifiable. He was started oral prednisolone and cyclophosphamide as induction therapy. He was doing good, discharged after a nine-day stay at the hospital and is currently under follow up.


